Biontech & MRNA: Revolution in der Pharmaindustrie
Ein Patent sagt gar nichts aus ob es auch funktioniert oder nicht.
Extrem gesagt kann eine Firma mit 100 Patenten ohne Erfolg dastehen während die andere Firma mit einem Patent, was zum Blockbuster wird, Mrd Umsätze generiert.
Außerdem finde ich es schwer einzuschätzen, was die Patente bringen, im Zweifel macht es ein Mitbewerber ähnlich und danach verklagen alle sich gegenseitig jahrelang...
Summit Therapeutics entwickelt sein Produkt schließlich in enger Zusammenarbeit mit dem chinesischen Akeso, die dann definitiv Fragen aufwerfen wird. Zwar entwickelt Biontech BNT327 auch mit einem Partner in China, aber kann halt selbst "im Westen" produzieren wofür Summit definitiv einen Partner brauchen würde der sich dann aber wiederum die Lizenzen in China erkaufen müsste, was dann erneut Fragen aufwerfen könnte.
Genau so spannend ist aber der Punkt, dass die Summit/Akeso-Studie wohl ausschließlich in China durchgeführt wird, was laut den bisherigen Statuten der Amerikaner wohl für eine Zulassung nicht ausreicht:
"At the same time, the industry consensus is that trials conducted in China will not reflect the racial diversity of the US population. Relying solely on Chinese-only studies like HARMONi-2, or trials conducted mainly in a country other than the US, may not support applications in the US, especially for major indications like NSCLC."
https://www.drugtimes.cn/2024/06/05/...appymerckexecutivescommentont/
Biontech hingegen hat seine Studie im Westen.
Am Ende könnte es darauf hinauslaufen, dass man aufgrund diverser Regelungen das Produkt von Summit erst noch in amerikanischer Studie durchführen muss oder man halt aufgrund diverser Sanktionsgefahr aufgrund des neuen Gesetzes nicht eventuell sowieso ausschließlich das Biontech Produkt kaufen müsste.
Um 11:15 kommt für biontech die letzte mit bnt113, der abstrakt dazu liest sich schonmal sehr gut:
Results
From 05/2017 to 01/2023, 17 patients with HPV16+ HNSCC participated in Arm 1A. 76% were male (n=13) with median age 63. Arm 1B enrolled 13 patients with anal (38%, n=5), HNSCC (23%, n=3), cervical (15%, n=2), other (23%, n=3) after failure of prior therapies; 77% were female (n=10) with median age 58. 19 (63%) received all 8 doses. Although no DLTs were reported, 2 patients in Arm 1A met individual MTDs. Grade≥1 ADRs ≤1wk after any vaccination/Grade≥3 ADRs ≤90d after last vaccination were experienced in 90% (n=26)/24% (n=7) patients among the 29 dosed. Disease control was seen in 5/7 patients (71%, 95% CI 29% – 96%) in Arm 1B with post-treatment tumour scans. Of evaluable patients, 70% mounted cellular immune responses against E6/7 as measured ex vivo by IFNγ ELISpot : 11/13 patients Arm 1A and 3/7 in Arm 1B. Anti-E7 IgG-Ab responses were detected in 8/15 (Arm 1A) and 5/9 (Arm 1B) patients. De novo T cell responses against E7 epitopes were found ex-vivo at a dose of 72.8 μg (Arm 1A Cohort II and Arm 1B); CD4+ and CD8+ E7-reactive T cell clones, were detected after in-vitro stimulation also at 29 μg (5/6 patients, Arm 1A). Vaccine-induced E7/E6-specific T cells reached up to 4.9% of CD8 T cells in circulation and were tracked by multimers in several patients longitudinally. TCR-seq (TIL and blood analysis) and TME characterisation data will be presented.
Conclusions
BNT113 was overall well tolerated. Vaccine-induced immune responses were seen in the majority of patients in the adjuvant setting and in the presence of advanced disease.
Es waren auch meine Überlegungen, dass (zumindest) zeitliche Vorsprung von Summit mit diesem Act eigentlich nicht mehr relevant ist.
Ich frage mich, warum Dermapharm heute (ohne Nachrichten) anspringt...
Optionen
| Boardmail an "Synoptic" |
Wertpapier: BioNTech SE ADR |
Quelle: https://optioncharts.io/options/BNTX/max-pain
Optionen
| Boardmail an "Nachdenker 2030" |
Wertpapier: BioNTech SE ADR |
Angehängte Grafik:
16-09-2024_10-55-20_temp_biontech.jpg (verkleinert auf 29%)
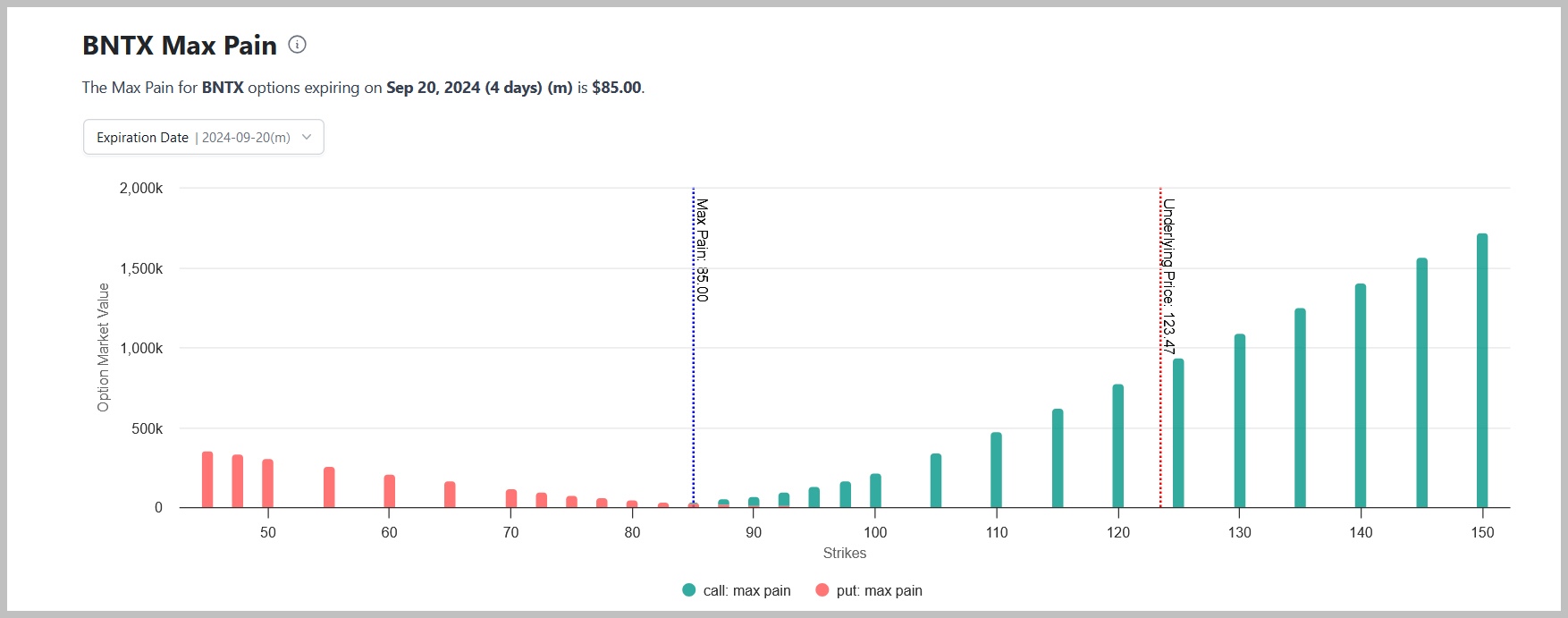
16-09-2024_10-55-20_temp_biontech.jpg (verkleinert auf 29%)
Nichtsdestotrotz hat auch das anvisierte Keytruda keine Monopolstellung, denn Opdivo hat mit 9 Milliarden Umsatz auch kein kleines Produkt am Markt.
Am Ende sind beide zugelassen und in verschiedenen Indikationen in der Anwendung und haben somit ihren Platz im Markt.
Also wird wohl realistisch gesehen Platz für zwei Mitstreiter im
Markt sein, alleine schon, weil die Anwendungen durch die verschiedenen Produkte sehr breit sein werden und auch immer mal jemand mit Produktionsengpässen zu tun haben wird.
Ob der Umsatz aber so hoch sein wird wie bei Keytruda und Opdivo (zusammen 34 Milliarden) darf dann auch bestritten werden, da kommt es dann auch darauf an wie viele Dosen über welchen Zeitraum verabreicht werden müssen, wie die Produktionskosten sind und was man im Endeffekt vor Allem im Markt durchsetzen kann.
https://investors.biontech.de/static-files/...-416e-8470-c465210101e5
Warum wohl?
HPV16-getriebene Kopf-Hals-Plattenepithelkarzinome (HNSCC), insbesondere:
(1) Oropharynxkarzinome (Krebs des Mundrachens)
(2) Zervixkarzinome (Gebärmutterhalskrebs)
(3) Analkarzinome Die machen in Mainz keine halben Sachen.
Quelle: ESMO-Präsentation Seite 74
https://investors.biontech.de/static-files/...-416e-8470-c465210101e5
(sehr gut! lesenswert!)
Weitere Wirksamkeitsdaten in der Datei und weitere Ausweitungen z.B. Seite 70.
Moderation
Zeitpunkt: 17.09.24 09:55
Aktion: Löschung des Beitrages
Kommentar: Fehlender Mehrwert für andere Forenteilnehmer
Zeitpunkt: 17.09.24 09:55
Aktion: Löschung des Beitrages
Kommentar: Fehlender Mehrwert für andere Forenteilnehmer
Optionen
| Boardmail an "Synoptic" |
Wertpapier: BioNTech SE ADR |
Die Frage bleibt für mich, wie viel die verkaufen wollen, ihr Bestand ist immer noch sehr hoch. Bin da bei dir Phoenix, an einem Tag wie gestern/vorgestern geht das "kursschonend". Ansonsten halt eher nicht. Wenn die bis auf 0 verkaufen wollen, nach und nach, warum auch immer, steht uns noch lange ein gebremster Kurs bevor...
https://investors.biontech.de/static-files/...-4be1-a6c1-d1d6d5869d5d
https://investors.biontech.de/static-files/...-41af-afb8-af855a743c46
Trotzdem kann man das, trotz hohem Handelsvolumen gestern, m.m.n. eleganter machen als plump ein 500ter block reinzuhämmern
Ansonsten belastet es halt solange, wie sie immer wieder solche Pakete raus werfen. Und das können sie halt noch oft...
Wir wissen halt nicht was die eigentlich vorhaben, schauen wir wo die Reise hin geht, hoffentlich hoch :)
Solch große Aktienpakete werden oft außerbörslich an Großinvestoren verkauft. In diesem Fall (so wie es aussieht) wurde kein Großinvestor gefunden und so werden die Stücke (kursschonend) auf den Markt geschmissen. Warum hat sich kein Großinvestor gefunden? Darüber dachte ich über das WE nach.
Optionen
| Boardmail an "Synoptic" |
Wertpapier: BioNTech SE ADR |
Meine persönliche Strategie (ohne Gewähr): Ich habe einen großen Teil von Biontech verkauft. Mein Ziel besteht darin, wieder günstig einzusteigen. Ob das richtig war oder nicht funktioniert, kann ich erst nach einiger Zeit berichten. Sicher bin ich mir nicht. Aber ich probier dies jetzt mal.
PS: Keine Anlageempfehlung. Ich könnte mich auch irren.
Optionen
| Boardmail an "Nachdenker 2030" |
Wertpapier: BioNTech SE ADR |
Falls sich während der zulassungsrelevanten Studie der Goldstandard bei dieser Indikation verändert. Wie ist hier die weitere Vorgehensweise?
Kann die Studie ohne Änderung fortgeführt werden? Und falls das in der Studie verwendete neue Arzneimittel den vormaligen Goldstandard übertrifft aber gegenüber dem neuen Goldstandard unterlegen wäre, dennoch die Zulassung erhalten?
Oder muss gegen den neuen Goldstandard eine Nichtunterlegenheit nachgewiesen werden und die Studie ist um einen Vergleichsarm mit dem neuen Goldstandard zu erweitern.
"In einer klinischen Studie, bei der die Nichtunterlegenheit eines neuen Arzneimittels gegenüber einem bestehenden Goldstandard nachgewiesen werden soll, könnte der Ablauf durch eine Änderung des Goldstandards während der laufenden Studie beeinflusst werden. Die Vorgehensweise in einem solchen Fall hängt von mehreren Faktoren ab, darunter regulatorische Anforderungen und die wissenschaftliche Beurteilung der Situation.
Hier sind die möglichen Szenarien und ihre Konsequenzen:
### 1. **Studie ohne Änderung fortsetzen**
- Wenn sich der Goldstandard während einer Studie ändert, kann es unter bestimmten Umständen möglich sein, die Studie ohne Anpassungen fortzuführen. Dies hängt davon ab, ob der ursprüngliche Goldstandard weiterhin als akzeptable Vergleichsmethode von den Zulassungsbehörden anerkannt wird. In diesem Fall könnte die Studie abgeschlossen werden, und der Vergleich mit dem vorherigen Goldstandard wäre weiterhin gültig, solange die Ergebnisse solide und relevant sind.
### 2. **Überlegenheit gegenüber dem alten Goldstandard, aber Unterlegenheit gegenüber dem neuen**
- Sollte das neue Arzneimittel den früheren Goldstandard übertreffen, aber dem neuen Goldstandard unterlegen sein, wäre die Zulassung schwieriger. Zulassungsbehörden könnten verlangen, dass das neue Arzneimittel zumindest eine **Nichtunterlegenheit** gegenüber dem neuen Standard nachweist. Falls dies nicht möglich ist, könnte die Zulassung in Frage gestellt werden, es sei denn, das Medikament hat einen spezifischen Vorteil (z. B. bessere Verträglichkeit oder einfachere Verabreichung), der die Unterlegenheit in der Wirksamkeit rechtfertigt.
### 3. **Anpassung der Studie und Hinzufügen eines neuen Vergleichsarms**
- In vielen Fällen fordern Zulassungsbehörden, dass der neue Goldstandard als Vergleichsarm in die Studie aufgenommen wird, um die **Nichtunterlegenheit** oder **Überlegenheit** auch gegenüber dem neuen Standard nachzuweisen. Dies könnte bedeuten, dass die Studie angepasst wird und zusätzliche Patienten in einen Arm mit dem neuen Goldstandard randomisiert werden. Solche Anpassungen erfordern oft neue Genehmigungen durch Ethikkommissionen und können die Studiendauer verlängern.
### Regulatorische Anforderungen
- Die Anforderungen können je nach Zulassungsbehörde (z. B. FDA in den USA, EMA in der EU) variieren. Diese Behörden bewerten oft die **wissenschaftliche Relevanz** der Studienergebnisse im Kontext der neuen Standards und berücksichtigen die Marktnachfrage sowie medizinische Notwendigkeiten. Sollte der neue Goldstandard deutliche Verbesserungen zeigen, könnte die Nichtunterlegenheit gegenüber dem neuen Standard eine Voraussetzung für die Zulassung werden.
Zusammenfassend hängt die Vorgehensweise stark von den spezifischen Anforderungen der jeweiligen Zulassungsbehörde und der medizinischen Relevanz der Studie ab. In vielen Fällen ist eine Anpassung der Studie erforderlich, wenn sich der Goldstandard während des Prozesses ändert, um die Marktzulassung zu sichern."